G-Proteine und ihre Signalwirkung im Herz-Kreislauf- und Immunsystem
Forschungsbericht (importiert) 2014 - Max-Planck-Institut für Herz- und Lungenforschung - W. G. Kerckhoff-Institut
Ziel einer Arbeitsgruppe am MPI für Herz- und Lungenforschung ist es, die G-Protein-vermittelten Signalwege im Herz-Kreislauf-System und im Immunsystem zu verstehen. Es sind vor allem die heterotrimeren G-Protein-Familien Gq/G11 und G12/G13, die hier intrazellulär vermitteln. Zu den Arbeiten gehört die Identifizierung neuer Rezeptoren, die diese Signalwege aktivieren, sowie die Aufklärung der nachgeschalteten intrazellulären Signalwege. Langfristiges Ziel dieser Arbeiten ist es, neue Zielmoleküle zur Behandlung chronischer Erkrankungen des kardiovaskulären Systems zu entdecken.
Der Fokus der Arbeitsgruppe um Nina Wettschureck lag in den letzten Jahren auf der Analyse G-Protein-vermittelter Signalwege in Endothelzellen, Kardiomyozyten (Herzmuskelzellen) und Immunzellen, wobei die G-Protein-Familien Gq/G11 und G12/G13 einen besonderen Schwerpunkt bildeten (Abb.1).


G-Proteine bei Herzmuskelverdickung und Herzschwäche
Nina Wettschureck und ihr Team konnten kürzlich nachweisen, dass G-Protein-aktivierte Signalwege eine zentrale Rolle bei der Entstehung von Herzhypertrophie (Herzmuskelverdickung) und Herzfibrose spielen [1; 2; 3]. Herzhypertrophie ist die physiologische Reaktion des Herzens auf anhaltende Druck- oder Volumenüberlastung, z. B. im Rahmen eines arteriellen Bluthochdruckes oder einer Erkrankung der Herzklappen. Chronische Herzhypertrophie und die damit einhergehende Fibrosierung des Herzens, bei der das Muskelgewebe steifer wird, ist mit einem erhöhten Risiko für Herzinsuffizienz (Herzschwäche) und Rhythmusstörungen verbunden. Forscher suchen daher nach neuen therapeutischen Ansätzen, um diese Veränderungen zu vermeiden.
Die Arbeitsgruppe in Bad Nauheim zeigte, dass die G12/13-Familie eine zentrale Rolle bei der Entstehung von Herzhypertrophie und -fibrose spielt [2], darüber hinaus konnte eine wichtige Funktion des Signalmoleküls RhoGEF12 nachgewiesen werden. Untersuchungen an Kardiomyozyten unter Druckbelastung zeigten, dass RhoGEF12 nicht nur durch die heterotrimeren G-Proteine der G12/13-Familie aktiviert wird, sondern auch durch β1-Integrine, die als Sensoren mechanischen Stresses dienen. Mäuse, bei denen RhoGEF12 in Herzmuskelzellen gezielt ausgeschaltet wurden, waren bei kardialer Drucküberlastung vor Hypertrophie und Fibrosierung geschützt; langfristig zeigten diese Tiere eine verbesserte Pumpleistung des Herzens und ein längeres Überleben (Abb. 2).
 und einer Maus mit Kardiomyozyten-spezifischer Inaktivierung von RhoGEF12 (\"cmc-Gef12-KO\") ein Jahr nach Induktion einer kardialen Drucküberlastung durch Transversale Aortenkonstriktion (TAC). Sham steht für Scheinoperation. Die Entwicklung einer Herzinsuffizienz im Kontrolltier ist an der Erweiterung des linken Ventrikels zu erkennen, sie ist in Knockout-Tieren wesentlich schwächer ausgeprägt.")

Aus klinischer Sicht ist ebenfalls bedeutsam, dass die Inaktivierung von RhoGEF12 auch bei Tieren mit bereits bestehender Herzwandverdickung zur Verbesserung des Krankheitsverlaufes führte. Diese Daten zeigen, dass RhoGEF12 aufgrund seiner zentralen Position als Integrator mechanischer und hormoneller Reize ein attraktives Ziel ist, um die Herzhypertrophie pharmakologisch zu beeinflussen. Der nächste Schritt ist daher, in Kooperation mit dem Lead Discovery Center der Max-Planck-Gesellschaft spezifische Inhibitoren für RhoGEF12 zu entwickeln.
Die G12/13-Familie in Endothelzellen
Untersuchungen in Zellkulturen und in genetisch veränderten Mäusen zeigten, dass G-Proteine der G12/13-Familie auch eine wichtige Rolle bei der Regulation der Expression des VEGF-Rezeptors spielen, eines zentralen Regulators des Gefäßwachstums [4]. Eine Hemmung dieses Signalweges in Endothelzellen führte zu einer Reduktion des Gefäßwachstums in Tumoren, was wiederum dazu führte, dass sich das Tumorwachstum verlangsamte. Eine pharmakologische Modulation des G12/13-vermittelten Signalweges in Endothelien könnte daher ein neuer Ansatz sein, um die pathologische Gefäßneubildung im Rahmen des Tumorwachstums zu beeinflussen.
Wirkmechanismus von Dimethylfumarat bei Multipler Sklerose
Ein weiterer Fokus des Teams ist die Identifizierung neuer Funktionen von G-Protein-gekoppelten Rezeptoren. In enger Kooperation mit der Arbeitsgruppe um Markus Schwaninger (Universität Lübeck) gelang es zu zeigen, dass die kürzlich zur Therapie der Multiplen Sklerose zugelassene Substanz Dimethylfumarat ihren therapeutischen Effekt durch den Rezeptor HCA2 vermittelt [5]. Bei der Multiplen Sklerose handelt es sich um eine chronisch entzündliche Erkrankung des zentralen Nervensystems, die auf einer autoimmunen Zerstörung der Myelin-Hüllen der Nervenfasern beruht. Seit einigen Jahren ist bekannt, dass die bislang zur Therapie der Schuppenflechte eingesetzte Substanz Dimethylfumarat auch bei Multipler Sklerose wirksam ist; welcher Mechanismus dieser Wirkung zugrunde liegt, war weitgehend unklar.

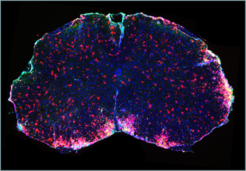
Die Arbeitsgruppe konnte nun zeigen, dass der protektive Effekt von Dimethylfumarat in einem Mausmodell der Multiplen Sklerose verloren geht, wenn den Tieren der G-Protein-gekoppelte Rezeptor HCA2 fehlt. Begleitende Untersuchungen in Zellkultursystemen beweisen, dass Dimethylfumarat die Migration und Adhäsion neutrophiler Granulozyten in einer HCA2- abhängigen Weise hemmt (Abb. 3). Basierend auf diesen Befunden ist nun die Entwicklung neuartiger Therapeutika für die Behandlung von Multipler Sklerose mit verbessertem Wirkungs- und Nebenwirkungsprofil das nächste Ziel.
Weitere Projekte
Ein aktueller Schwerpunkt der Arbeitsgruppe ist die Untersuchung der Rolle Gq/11-vermittelter Signaltransduktionswege bei der Regulation endothelialer Permeabilität und postnataler Angiogenese. Auch die Rolle G-Protein-vermittelter Signalwege in Immunzellen ist weiterhin ein wichtiges Thema, insbesondere im Rahmen der Multiplen Sklerose. Im kardialen Bereich liegt der Fokus der Arbeitsgruppe momentan auf den Mechanismen der Aktivierung und der Inaktivierung kardialer Fibroblasten bei Herzhypertrophie und Myokardinfarkt. Basierend auf diesen Untersuchungen ist es langfristig das Ziel, innerhalb G-Protein-abhängiger Signalkaskaden Zielstrukturen zu identifizieren, die eine Zelltyp-spezifische pharmakologische Beeinflussung pathophysiologischer Prozesse ermöglichen.
Literaturhinweise
The Journal of Experimental Medicine 210, 665-73 (2013)
DOI: 10.1084/jem.20122126Circulation 126, 1972-1982 (2012)
DOI: 10.1161/CIRCULATIONAHA.112.109256Nature Medicine 7, 1236-1240 (2001)
DOI: 10.1038/nm1101-1236Developmental Cell 25, 427-434 (2013)
DOI: 10.1016/j.devcel.2013.04.008
The Journal of Clinical Investigation 124, 2188-2192 (2014)
DOI: 10.1172/JCI72151